Роль и практика фармаконадзора в российском здравоохранении
Современные лекарственные средства (ЛС) в настоящее время широко используются во всех областях медицины. Их применение позволяет существенно повышать качество жизни пациентов, улучшать прогноз и снижать смертность при многих заболеваниях. С другой стороны, внедрение в клиническую практику инновационных препаратов с высокой биологической активностью, растущая сенсибилизация населения к биологически активным и химическим веществам, нерациональное использование ЛС, полипрагмазия, медицинские ошибки, присутствие на фармацевтическом рынке большого количества дженериков, часть из которых не соответствует критериям качества, повысили риски развития нежелательных реакций (НР) при лекарственной терапии. Имеющиеся данные показывают, что НР являются частой причиной госпитализации, требуют дополнительного лечения и даже могут приводить к смерти пациентов [1–6]. Сегодня применение ЛС в клинической практике базируется на обязательной оценке соотношения «польза/риск», когда вероятная польза от применения ЛС существенно перевешивает потенциальный риск. Это требует не только убедительных доказательств эффективности лекарственных средств, но и изучения их безопасности. Наблюдение за безопасностью ЛС осуществляется в рамках системы фармаконадзора. По определению ВОЗ «фармаконадзор (Pharmacovigilance, vigilance – бдительность, англ.) – это научные исследования и виды деятельности, связанные с выявлением, оценкой, пониманием и предотвращением нежелательных реакций или любых других проблем, связанных с лекарственным препаратом» [7].
Этапы развития фармаконадзора
Становление фармаконадзора в мире было связано с рядом лекарственных трагедий [8, 9]. Так, в 1937 г. в США 107 человек (преимущественно дети) умерли при применении поступившей на рынок жидкой формы сульфаниламидов (торговое наименование «Элексир»), где в качестве растворителя производитель использовал диэтиленгликоль. О токсичности этого вещества в то время было уже известно, однако фармацевтический химик компании-производителя об этом ничего не знал. После произошедшего случая в 1938 г. в Федеральный закон США «Food, Drug and Cosmetic Act» было добавлено требование доказательства производителем безопасности нового лекарственного препарата (ЛП) до его выхода на рынок.
Ключевое событие в истории фармаконадзора, которое получило название «талидомидовая трагедия», произошло в 1958—1961 гг. и было связано с широким применением беременными женщинами для лечения токсикоза в I триместре нового препарата талидомида, считавшегося в то время безопасным и обладавшим седативным, снотворным и противорвотным действием. В конце 1961 года, почти в одно время, профессор Ленц в Германии и доктор Макбрайд в Австралии выявили связь между возросшим числом врожденных пороков у новорожденных и тем фактом, что матери этих детей принимали талидомид на ранних сроках беременности. Всего, по различным оценкам, в результате применения талидомида от 8000 до 12 000 детей родились с физическими уродствами (наиболее разрушительный удар пришелся на Западную Германию – более 4000 пострадавших). Около 5000 детей не погибли в раннем возрасте, а остались инвалидами на всю жизнь, в основном в связи с развитием редкой врожденной патологии – фокомелии (буквально «ласты тюленя» – недоразвитие проксимальных отделов конечностей).
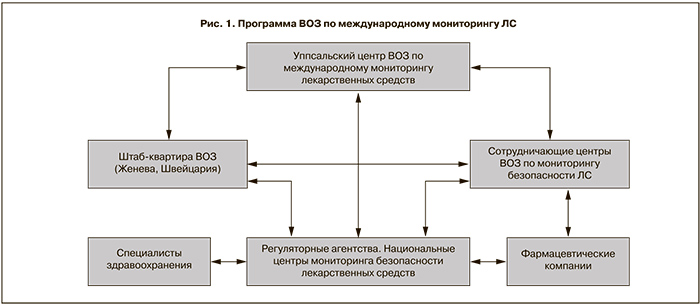
Талидомидовая трагедия послужила мощным толчком к развитию фармаконадзора в мире. Данные события привели к принятию в 1962 г. в США поправок к «Закону о ЛС» (Drug Amendment Act), требующих по существу проведения производителями предрегистрационных исследований с целью доказательства не только безопасности, но и эффективности нового ЛП, что в последующем стало обычной практикой в большинстве стран мира. Талидомидовая трагедия также стимулировала развитие системы спонтанных сообщений (СС) как основного на тот момент способа получения информации о нежелательных реакциях (НР) лекарственных средств. Метод спонтанных сообщений определяется как добровольное или в соответствии с законодательными требованиями информирование специалистами здравоохранения соответствующих регуляторных структур о выявляемых НР. Впервые это требование было введено в Германии в 1963 г., затем в Великобритании в 1964 г. В Великобритании указанный метод получил название системы «желтой карты» (по цвету бланка-извещения). Были разработаны основные принципы метода и практические аспекты применения, что привело к его широкому и эффективному использованию в стране [10, 11]. С 1967 г. действует программа ВОЗ по международному мониторингу безопасности ЛС (рис. 1). В рамках этой программы в 1968 г. в городе Уппсала (Швеция) открыт Центр сотрудничества ВОЗ по международному мониторингу лекарств (UMC) (http://www.who-umc.org/.)
Становление системы фармаконадзора в СССР [1] относится к 1969 г., когда был создан Отдел учета, систематизации и экспресс-информации о побочном действии ЛС, который в 1973 г. был преобразован во Всесоюзный организационно-методический центр по изучению побочных действий лекарств (ВЦПДЛ). Данный центр в целом выполнял функции, которые возлагаются на систему мониторинга безопасности ЛС. Стоит отметить, что именно в этот период была создана первая специальная форма карты-извещения о НР. Два раза в год выходили информационные письма, в которых рассматривались все зарегистрированные нежелательные реакции на медицинские препараты, издавался ежемесячный реферативный журнал «Побочные действия лекарственных средств». В 1991 г. при ликвидации Минздрава СССР работа по выявлению и регистрации НР была приостановлена. Лишь в 1997 г. на базе кафедры общей и клинической фармакологии Российского университета дружбы народов (РУДН) под руководством чл.-корр. РАМН В.К. Лепахина был образован Федеральный центр по изучению побочных действий лекарств, который в дальнейшем после создания в 2004 г. Федеральной службы по надзору в сфере здравоохранения и социального развития (далее Росздравнадзор) был преобразован в Федеральный центр мониторинга безопасности ЛС (ФЦ МБЛС) при ФГУ «Научный центр экспертизы средств медицинского применения» (ФГУ НЦЭСМП) Росздравнадзора. Важным моментом в этот период явилось формирование сети региональных центров мониторинга безопасности ЛС (РЦ МБЛС), выход в 1998 г. Федерального закона «О лекарственных средствах» №83-ФЗ, где на законодательном уровне была закреплена система СС. Была утверждена новая «Карта-извещение о побочном действии, нежелательной реакции или отсутствии ожидаемого терапевтического эффекта лекарственного средства», обеспечена доступность этой карты и рекомендовано ее наличие в каждом стационарном и амбулаторном учреждении. Трудно переоценить работу, которая была проведена Росздравнадзором совместно с ФЦ МБЛС по разработке единых всероссийских баз данных – автоматизированных информационных систем (АИС): подсистемы «Фармаконадзор» и «Мониторинг клинических исследований лекарственных средств» («МКИЛС») АИС Росздравнадзора, которые начали функционировать в 2008 г.
Нормативная правовая база
С 2010 г. в Российской Федерации произошли существенные изменения в нормативной правовой базе, регламентирующей работу в сфере фармаконадзора [12, 13]. Основным документом, который определил приоритет государственного контроля безопасности и эффективности ЛС, зарегистрированных на территории России, в настоящее время является Федеральный закон от 12.04.2010 №61-ФЗ «Об обращении лекарственных средств». Глава 13 этого закона полностью посвящена мониторингу безопасности лекарственных препаратов (ЛП), находящихся в обращении на территории РФ.
В соответствии со ст. 64 этой главы: ЛП, находящиеся в обращении на территории РФ, подлежат мониторингу безопасности в целях выявления возможных негативных последствий их применения, предупреждения пациентов и их защиты от применения таких препаратов.
Мониторинг безопасности ЛП осуществляется уполномоченным федеральным органом исполнительной власти на всех этапах их обращения на территории РФ.
Субъекты обращения ЛС обязаны сообщать в установленном уполномоченным федеральным органом исполнительной власти порядке обо всех случаях побочных действий, не указанных в инструкции по применению ЛП, о серьезных нежелательных реакциях, непредвиденных нежелательных реакциях при применении ЛП, об особенностях взаимодействия ЛП, которые были выявлены при проведении клинических исследований и применении ЛП.
За несообщение или сокрытие сведений, предусмотренных частью 3 настоящей статьи, лица, которым они стали известны по роду их профессиональной деятельности, несут ответственность в соответствии с законодательством РФ.
Постановлением Правительства Российской Федерации от 20.08.2010 №650 «О внесении изменений в некоторые акты Правительства Российской Федерации в связи с принятием Федерального закона «Об обращении лекарственных средств» государственная функция по проведению мониторинга безопасности ЛП, находящихся в обращении на территории Российской Федерации, возложена на Федеральную службу по надзору в сфере здравоохранения и социального развития. Росздравнадзор осуществляет сбор, обработку и анализ сведений по безопасности ЛП и направляет их в Министерство здравоохранения Российской Федерации (Департамент государственного регулирования обращения ЛС) для рассмотрения вопроса о возможности принятия административных решений (например, внести изменения в инструкцию по применению ЛП, приостановить применение или возобновить применение ЛП). Решения Минздрава России публикуются на сайте http://grls.rosminzdrav.ru/. На официальном сайте Росздравнадзора также доступна информация об изменении профиля безопасности ЛС, представленная в соответствующих информационных письмах.
По результатам мониторинга безопасности ЛП Федеральным законом №61-ФЗ предусмотрена возможность приостановления применения ЛП (ст. 65) и даже отмена государственной регистрации (ст. 32). В главе 6 (ст. 29) указано, что для подтверждения государственной регистрации ЛП требуется предоставить результаты мониторинга его безопасности. Эти новые законодательные требования повысили ответственность производителей и держателей регистрационных удостоверений за организацию системы изучения безопасности выпускаемых ими препаратов.
Следует отметить, что Российская Федерация, наряду со всеми странами, в настоящее время входящими в Таможенный союз, с 1998 г. является официальным участником международной программы ВОЗ по мониторингу безопасности ЛС, поэтому многие аспекты деятельности по выявлению, анализу и оценке безопасности ЛП в нашей стране проходят постоянный процесс гармонизации с международными стандартами фармаконадзора, вырабатываются единые правила мониторинга НР в рамках стран Таможенного союза.
Принципы фармаконадзора
Важным принципом фармаконадзора является изучение безопасности ЛП в процессе всего его «жизненного цикла», начиная с лабораторного тестирования, доклинических исследований на животных, предрегистрационных клинических исследований (КИ) и заканчивая всем периодом его обращения на рынке [1, 14, 15]. Оценка безопасности ЛП в предрегистрационных клинических исследованиях ограничена небольшим числом пациентов (3500–5000), непродолжительным периодом лечения (1,5–2 года), исключением некоторых групп (дети, беременные, пожилые). Международная практика показывает, что к моменту выхода препарата на рынок удается выявить лишь около 50% его нежелательных реакций [15–17]. Как правило, это частые реакции (частота развития от 1 до 10%), обусловленные фармакологическими свойствами нового ЛП. Таким образом, для получения полной информации о рисках, связанных с применением ЛС, требуется постоянный мониторинг их безопасности в постмаркетинговом периоде. На этом этапе возможно выявление редких и очень редких НР (частота встречаемости от 0,1% и ниже), изучение безопасности ЛС в особых группах пациентов (пожилые, дети, беременные и кормящие грудью матери), выяснение факторов риска развития НР, продолжение изучения неблагоприятных взаимодействий ЛС, а также влияния ЛП на прогноз заболевания и уровень смертности [18]. Очень важно, чтобы в сборе информации о лекарственных препаратах принимали участие все заинтересованные лица (представители официальных регуляторных органов, фармацевтических компаний, производители ЛС, специалисты здравоохранения и пациенты), и было правильно выстроено их взаимодействие [19].
К настоящему времени в мире разработано достаточно много методов выявления нежелательных реакций на ЛС. Однако основным методом фармаконадзора во всех странах, включая Российскую Федерацию, является метод СС. Метод СС, как и любой другой метод, имеет свои преимущества и ограничения [12, 14, 17, 20–22]. Он позволяет получать информацию обо всех препаратах, используемых на рынке в реальных условиях, без ограничения периода наблюдения, у всех групп больных. Возможно выявление очень редких и непредвиденных НР. К недостаткам метода относят низкий показатель регистрации НР (примерно 10% от выявляемых, даже в странах с хорошо налаженной системой), получение СС низкого качества с неполной и неточной информацией, невозможность определить частоту встречаемости НР, выявить отсроченные (например, тератогенные или канцерогенные) эффекты. Следовательно, полученная информация о новых рисках часто требует проверки в специальных исследованиях. В свое время с помощью метода СС удалось выявить теперь хорошо известные НР: удлинение интервала QT при приеме цизаприда и астемизола, «серый синдром новорожденных» при приеме левомицетина, агранулоцитоз при приеме фенилбутазона, тромбоэмболии при приеме оральных контрацептивов, поражение клапанов сердца при приме фенфлурамина, нарушение полей зрения при приеме вигабатрина и др.
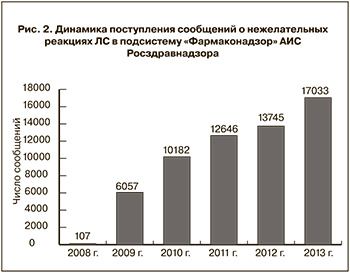 Главная задача метода СС – это выявление сигнала безопасности. Термин «сигнал» в фармаконадзоре определяется как «информация о возможной причинно-следственной связи между НР и приемом ЛС, о которой ранее не было ничего известно или сведения были недостаточно информативными» [23]. Сигналом может быть еще неизвестная НР или новая информация об уже известной реакции. Для успешной работы метода СС требуется определенная активность сообщающих и достаточная мощность базы (т.е. количество сообщений). Так, критериями эффективности метода считается поступление в структуры фармаконадзора 100 сообщений на 1000 врачей в год (показатель, характерный для Великобритании, Канады, Швеции, Австралии, Новой Зеландии и некоторых других стран), или примерно 600 сообщений на 1 млн жителей (рекомендовано ВОЗ в 2012 г.) [24]. При этом минимальным показателем, при котором метод СС работает, считается 100 сообщений на 1 млн жителей. В мире существует много национальных и объединенных баз СС, которые содержат уже миллионы таких сообщений, например, международная Уппсальская база НР (WHOdatabase –Vigibase) включает сегодня более 9 млн сообщений [25] . Для сравнения приводим данные по АИС Росздравнадзора (рис. 2).
Главная задача метода СС – это выявление сигнала безопасности. Термин «сигнал» в фармаконадзоре определяется как «информация о возможной причинно-следственной связи между НР и приемом ЛС, о которой ранее не было ничего известно или сведения были недостаточно информативными» [23]. Сигналом может быть еще неизвестная НР или новая информация об уже известной реакции. Для успешной работы метода СС требуется определенная активность сообщающих и достаточная мощность базы (т.е. количество сообщений). Так, критериями эффективности метода считается поступление в структуры фармаконадзора 100 сообщений на 1000 врачей в год (показатель, характерный для Великобритании, Канады, Швеции, Австралии, Новой Зеландии и некоторых других стран), или примерно 600 сообщений на 1 млн жителей (рекомендовано ВОЗ в 2012 г.) [24]. При этом минимальным показателем, при котором метод СС работает, считается 100 сообщений на 1 млн жителей. В мире существует много национальных и объединенных баз СС, которые содержат уже миллионы таких сообщений, например, международная Уппсальская база НР (WHOdatabase –Vigibase) включает сегодня более 9 млн сообщений [25] . Для сравнения приводим данные по АИС Росздравнадзора (рис. 2).
Среднее число СС на 1 млн населения РФ в 2013 г. составило 96.
Следует отметить, что за 5 лет (база начала функционировать в конце 2008 г.) количество ежегодно поступающих в Росздравнадзор сообщений постоянно увеличивается, однако остается по-прежнему существенно ниже, чем в Европейском Союзе или в США. Только в 2013 г. количество сообщений на 1 млн жителей страны приблизилось к минимальному эффективному уровню, но при этом эксперты, работающие с АИС, отмечают, что приходит довольно много сообщений ненадлежащего качества. Общее количество СС в базе «Фармаконадзор» на конец 2013 г. достигло 60 тыс. Приведенные данные показывают, что российская национальная база НР сегодня не может функционировать эффективно, в частности, активно выявлять новые риски применения отечественных ЛС. К тому же для подтверждения новой информации о безопасности зарубежных ЛС требуется обращение к более крупным базам (например, Vigibase). Таким образом, для обеспечения эффективного и безопасного лечения пациентов на территории нашей страны необходимо дальнейшее совершенствование метода СС, что требует совместных усилий всех заинтересованных сторон и, прежде всего, сотрудников практического здравоохранения.
Мониторинг на уровне медицинской организации
Важно понимать, что первыми встречаются с нежелательными реакциями на препарат врачи, реже фармацевты [26–28]. Поэтому остановимся более подробно на практических аспектах организации мониторинга безопасности ЛП на уровне лечебно-профилактических учреждений (ЛПУ). Прежде всего, необходимо, чтобы каждый врач рассматривал работу по выявлению нежелательных реакций на ЛП, правильную их регистрацию и информирование соответствующих регуляторных органов как важную профессиональную обязанность, закрепленную Федеральным Законом № 61-ФЗ (гл. 13, ст. 64, пп. 3, 4). Полезно знать два основных международных принципа метода СС. Во-первых, сообщающий не должен решать, точно ли подозреваемый препарат вызвал данную НР, необходимо лишь предполагать наличие хотя бы минимальной причинно-следственной связи «ЛП-НР» [29]. Во-вторых, СС о НР не может быть основанием для наказания и преследования врача (в частности, не может быть использовано при возникновении судебных исков в связи с врачебными ошибками). Безусловно, что многие НР не являются результатом именно врачебной ошибки [30].
Для правильной организации системы фармаконадзора на уровне ЛПУ необходимо начать с назначения ответственного специалиста по мониторингу безопасности ЛП. Как правило, таким специалистом сегодня является врач – клинический фармаколог (Приказ Минздрава России от 05.05.1997 №131 «О введении специальности «Клиническая фармакология»). Современная система обучения клинических фармакологов позволяет именно этим специалистам наилучшим образом осуществлять информирование и обучение медицинского персонала по вопросам фармаконадзора [31, 32]. На первом этапе часто требуется проведение серьезной разъяснительной работы среди врачей о значении СС о НР для повышения безопасности лекарственной терапии и улучшения качества оказания медицинской помощи. Далее клинический фармаколог помогает врачам выявлять НР, выяснять все условия их развития (иногда для этого требуется провести дополнительные исследования, в частности, определение концентрации подозреваемого препарата в крови), правильно регистрировать НР в истории болезни/амбулаторной карте, заполнять соответствующую карту-извещение.
Метод СС и современное российское законодательство требует информирование специалистами здравоохранения соответствующих регуляторных органов (в Российской Федерации – Росздравнадзора) о выявляемых НР на ЛП. На первых этапах организации системы фармаконадзора в ЛПУ часто советуют составлять отчеты обо всех случаях НР. Это важно для выработки у врачей «культуры сообщений». В последующем, безусловно, следует сообщать в регуляторные органы о НР, перечисленных в ст. 64 п. 3 Федерального закона от 12.04.2010 №61-ФЗ «Об обращении лекарственных средств». К ним относятся непредвиденные и серьезные НР, а также неблагоприятные эффекты лекарственного взаимодействия. Определения непредвиденной и серьезной нежелательной реакции приводится в ст. 4 указанного Федерального закона.
Непредвиденная нежелательная реакция – ответ организма, сущность и тяжесть которого не соответствуют информации, изложенной в инструкции по применению ЛП. Эталонной информацией о ЛП для врача является действующая инструкция (часто с внесенными изменениями) по применению ЛП, доступная на сайте: http://grls.rosminzdrav.ru. Если в данной инструкции не описана выявленная НР или указана другая тяжесть, частота или характер, то такая НР расценивается как непредвиденная и подлежит сообщению в Росздравнадзор.
Серьезная нежелательная реакция – ответ организма, связанный с применением ЛП, приведший к смерти, врожденным аномалиям или порокам развития, либо представляющий собой угрозу жизни. Этот вид НР требует госпитализации или приводит к стойкой утрате трудоспособности и/или инвалидности. Необходимо различать понятия «серьезная НР» и «тяжелая НР»: серьезная НР не всегда бывает тяжелой, а тяжелая – серьезной.
Кроме этого Росздравнадзор в своих методических рекомендациях настоятельно рекомендует информировать его:
- о всех нежелательных реакциях на новые ЛС (применение на рынке менее 5 лет);
- о случаях применения на фоне беременности ЛС, противопоказанных при беременности;
- об эпизодах неэффективности ЛС, связанных со значительной угрозой жизни или здоровью человека (препараты для лечения угрожающих жизни заболеваний, вакцины, контрацептивы и др.);
- о случаях передозировки, ставших причиной серьезных НР, а также о случаях злоупотребления лекарственным препаратом (в т.ч. возникновение лекарственной зависимости);
- о случаях использования препаратов в целях причинения вреда жизни и здоровью человека;
- о случаях, в которых потенциально подозревается несоответствие качества ЛП нормативной документации.
Сроки сообщения в Росздравнадзор установлены приказом Министерства здравоохранения и социального развития Российской Федерации 26.08.2010 №757н: не позднее 15 календарных дней со дня, когда стала известна соответствующая информация. О летальных реакциях на препарат рекомендовано информировать Росздравнадзор в течение 24 часов после получения сведений (Информационное письмо Росздравнадзора от 11.04.2012 №04И-266/12).
Способы предоставления информации о подозреваемой НР в Росздравнадзор подробно описаны в информационном письме Росздравнадзора от 02.04.2012 №04И-232/12 «По предоставлению сведений о нежелательных реакциях на ЛП»:
- Заполнение утвержденной карты «Извещение о побочном действии, НР или отсутствии ожидаемого терапевтического эффекта лекарственного средства», размещенной на интернет-сайте Росздравнадзора (рубрика «Лекарственные средства», подрубрика «Мониторинг безопасности лекарственных средств», «Карта-извещение»). Карта может быть распечатана и заполнена «от руки», но желательно заполнять ее в электронном виде. Далее карта может быть направлена в Росздравнадзор в отдел мониторинга эффективности и безопасности медицинской продукции по почте на адрес: 109074, г. Москва, Славянская площадь, д. 4, стр. 1; по факсу (8(495)-689-25-73) или по электронной почте pharm@roszdravnadzor.ru.
- Дистанционно через интернет напрямую в подсистему «Фармаконадзор» АИС Росздравнадзора. В настоящее время этот способ рассматривается как предпочтительный и более удобный. Теоретически доступ к АИС при желании может получить любой специалист здравоохранения, но в настоящее время наиболее распространена практика, когда Росздравнадзор предоставляет для каждого ЛПУ один доступ к электронной базе, который получает ответственный специалист за мониторинг безопасности ЛС в данном учреждении. Для получения персонифицированного доступа в АИС следует отправить со своего e-mail запрос по адресу pharm@roszdravnadzor.ru c информацией о фамилии, имени, отчестве уполномоченного по фармаконадзору и реквизитами организации. Логин и пароль придет на электронную почту, указанную в запросе.
Электронная карта-извещение, которая заполняется в АИС Росзравнадзора, имеет свои особенности, например, название подозреваемого препарата и описание НР вводятся не вручную, а выбираются из встроенных справочников. Более подробно с правилами заполнения всех пунктов извещения можно ознакомиться, изучив прилагаемую инструкцию. На текущий момент специалист, зарегистрированный в АИС, имеет доступ только к своим сообщениям с возможными комментариями эксперта Росздравнадзора.
Выше уже обсуждалась проблема важности количества сообщений для эффективной работы метода СС. Однако международная практика показывает, что хотя «недосообщение» о НР является слабым звеном данного метода, но все-таки качество этих сообщений важнее их количества [25, 33, 34].
Эксперты Росздравнадзора выделяют валидные сообщения (т.е. те, которые пригодны для дальнейшего анализа и оценки). Валидные сообщения должны содержать минимальную информацию, которая позволяет:
- идентифицировать отправителя (источник) сообщения;
- идентифицировать пациента (по инициалам, дате рождения, возрастной группе или полу);
- установить лечебный препарат, с которым предположительно связана НР;
- установить подозреваемую НР.
Не относятся к валидным сообщениям следующие случаи:
- если сообщающий сделал четкое, аргументированное заявление об отсутствии причинно-следственной связи «ЛС-НР» и эксперт подтверждает эту оценку;
- если сообщается только исход или следствие (например, пациент умер, госпитализирован или произведен аборт после 12 недель беременности по медицинским показаниям без дополнительной информации). Исключением являются случаи внезапной смерти, которые рассматриваются как подозреваемая НР.
Невалидные СС вносятся в базу данных, и дальнейшая работа с ними заключается в попытках получить недостающую для проведения анализа информацию.
Для качественного сообщения характерно полное и профессиональное заполнение всех граф извещения. В разделе «Значимая дополнительная информация» необходимо предоставить достаточную информацию об анамнезе и диагнозе пациента, условиях развития НР. Часто требуется указать результаты лабораторных и инструментальных исследований, а в случае смерти пациента привести описание аутопсии. В разделе «Другие лекарственные средства» предоставить данные о сопутствующей терапии/препаратах. Хотя оценка причинно-следственной связи «ЛС-НР» проводится на всех уровнях работы со СС, всегда остается важна ее первоначальная оценка врачом, поэтому не следует пропускать эту графу в сообщении. Для оценки причинно-следственной связи врач может полагаться на свой профессиональный опыт или воспользоваться алгоритмами, приведенными в соответствующих методических рекомендациях Росздравнадзора (доступны на официальном сайте). Поскольку сроки информирования Росздравндзора о выявляемых НР ограничены, особенно при летальном исходе для пациента в результате развившейся реакции, часто это не позволяет врачу сразу предоставить данные в полном объеме. В этом случае посылается первичное СС, которое должно содержать минимальную, указанную выше информацию. Далее при получении важной медицинской информации, которая может повлиять на оценку случая, причинно-следственную связь, указать исход, в АИС Росздравнадзора вводится повторное сообщение (их может быть несколько и специально разработанная кодировка позволяет идентифицировать все эти сообщения как относящиеся к одному случаю). Практика работы экспертов показывает, что врачи ЛПУ гораздо реже, чем сотрудники фармацевтических компаний, присылают повторные сообщения, что сказывается на возможности качественной оценки случаев НР.
Результаты мониторинга безопасности препарата в конкретном ЛПУ должны активно использоваться клиническим фармакологом для целенаправленного консультирования и обучения врачей и среднего медицинского персонала [35, 36]. Знания рисков, связанных с применением ЛП, позволяет выработать у врачей умение прогнозировать развитие НР, а значит и возможность их предупреждать, в том числе в результате рационального использования лечебных препаратов и их комбинаций [37]. Однако не все НР можно предупредить, поэтому важным моментов является умение медицинского персонала купировать НР в случае их развития (особое внимание следует уделить лечению таких серьезных НР как анафилактический шок, тяжелые анафилактоидные реакции, тяжелые аритмии, злокачественный гипертермический синдром и др.). В задачи клинического фармаколога входит и информирование медицинского персонала о наиболее важных результатах работы регуляторных органов отечественного и международного фармаконадзора, в частности о принятых решениях об отзыве препаратов с рынка или введении ограничений на их использование в РФ и других странах. Результаты мониторинга применяемых ЛС также позволяют вносить изменения в формулярный список ЛПУ и корректировать политику по закупкам медикаментов. В целом комплекс перечисленных мероприятий, как показывает практика, приводит к снижению частоты развития НР и более безопасному лечению пациентов в учреждениях здравоохранения [32].
Заключение
В разработанном Минздравом России документе «Стратегия лекарственного обеспечения населения Российской Федерации на период до 2025 года» представлены основные направления развития службы фармаконадзора, которые в т.ч. включают дальнейшее совершенствование общероссийской базы данных о нежелательных побочных действиях ЛС, введение процедуры оперативного изменения статуса ЛС (приостановление/отзыв регистрационного удостоверения) и внесение изменений в стандарты медицинской помощи при выявлении серьезных и (или) непредвиденных побочных эффектов; организацию системы постоянного мониторинга проведения клинических исследований на территории Российской Федерации на предмет выявления НР на ЛС; оперативное информирование медицинских работников о выявленных побочных действиях ЛС и изменении профиля безопасности лекарств (посредством Интернет-ресурсов, медицинских периодических изданий и т.д.). Данные положения становятся особенно актуальными в условиях проводимого реформирования отечественной фарминдустрии, которое предусматривает замещение импортных ЛС отечественными аналогами и разработку собственных оригинальных препаратов. Оценить риски, связанные с их применением, безопасность и соотношение «польза/риск» можно будет только при эффективном функционировании национальной российской системы фармаконадзора на всех уровнях, включая мотивированную и активную работу в этом направлении клинических фармакологов, врачей всех других специальностей, а также фармацевтов.
Нормативные правовые акты, методические рекомендации и информационные письма, затрагивающие вопросы организации мониторинга безопасности лекарственных средств в медицинских организациях
- Федеральный закон от 12.04.2010 № 61-ФЗ «Об обращении лекарственных средств», ст. 64.
- Приказ Минздравсоцразвития России от 26.08.2010 № 757н «Об утверждении порядка осуществления мониторинга безопасности лекарственных препаратов для медицинского применения, регистрации побочных действий, серьезных нежелательных реакций, непредвиденных нежелательных реакций при применении лекарственных препаратов для медицинского применения» (Зарегистрирован Минюстом России 31.08.2010 № 8324).
- Приказ Минздрава России от 19.06.2003 № 266 «Об утверждении Правил клинической практики в Российской Федерации» (зарегистрирован Минюстом России 20.06.2003 №4808).
- Приказ Минздрава России от 13.02.2013 № 66 «Об утверждении Стратегии лекарственного обеспечения населения Российской Федерации на период до 2025 года и плана ее реализации».
Документы, утвержденные Росздравнадзором:
- Методические рекомендации «Определение степени достоверности причинно-следственной связи «Неблагоприятная побочная реакция – лекарственное средство» (классификация и методы)» (утверждены 05.10.2008).
- Методические рекомендации по осуществлению управлениями Росздравнадзора по субъектам Российской Федерации государственной функции по мониторингу безопасности лекарственных препаратов, находящихся в обращении на территории Российской Федерации (утверждены 16.01.2012).
- Информационное письмо от 02.04.2012 № 04И-232/12 «О предоставлении сведений о нежелательных реакциях на лекарственные препараты».
- Информационное письмо от 11.04.2012 № 04И-266/12 «О срочном предоставлении сведений о летальных нежелательных реакциях на лекарственные препараты».
- Информационное письмо от 18.03.2013 № 16 И-261/13 «О мониторинге безопасности лекарственных средств в клинических исследованиях».


