В настоящее время рак шейки матки (РШМ) является одной из самых актуальных проблем онкогинекологии. Заболевание занимает третье место среди всех злокачественных новообразований у женщин в мире и первое по частоте среди женщин 15–39 лет, являясь самой частой причиной смерти от онкогинекологических заболеваний в развивающихся странах [1]. По данным ВОЗ ежегодно в мире регистрируется около 470 000 новых случаев рака шейки матки, и, несмотря на проводимые лечебные мероприятия, 233 000 больных умирают от этого заболевания. В 15% случаев РШМ диагностируется у женщин в возрастном диапазоне от 20 до 34 лет.
В структуре онкозаболеваемости женщин в Российской Федерации наибольший удельный вес имеют злокачественные новообразования репродуктивной системы (39,2%), при этом 18,3% – это опухоли половых органов. На рис. 1 представлена динамика постепенного роста заболеваемости РШМ с 2003 по 2013 гг. Удельный вес РШМ в структуре достигает 26,07%, что свидетельствует о недостаточности мероприятий по профилактике данного заболевания.
Несмотря на применение различных методов лечения,его результаты остаются неудовлетворительными. Так, в 2013 г. было зарегистрировано 15 427 новых случаев РШМ, при этом умерло 6522 женщины, что соответствует примерно 40%. Данные по возрастной стратификации смертности при РШМ представлены на рис. 2 [2].
 Развитию РШМ предшествуют изменения морфологии клеток эпителия шейки матки разной степени выраженности – так называемые цервикальные интраэпителтальные неоплазии (ЦИН1-3).
Развитию РШМ предшествуют изменения морфологии клеток эпителия шейки матки разной степени выраженности – так называемые цервикальные интраэпителтальные неоплазии (ЦИН1-3).
При этом ЦИН сопровождается нарушением дифференцировки клеток, утратой их полярности и появлением атипических клеток. В соответствии с гистологической терминологией, предложенной в 1950-х годах J.W. Reagan и соавт. [3], предопухолевые изменения шейки матки классифицировали как слабо выраженную дисплазию I степени (базальные клетки с атипией занимают менее трети эпителиального пласта), умеренно выраженную – II степени (базальные клетки с атипией занимают 2/3 эпителия), резко выраженную – III степени (патологические изменения захватывают всю толщу эпителия, нет деления на слои) и внутриэпителиальный рак (рак in situ). В 1960-х годах R.M. Richard [4] предложил термин «цервикальная интраэпителиальная неоплазия». В 1968 г. появилась новая классификация, где ЦИН 1 и ЦИН 2 соответствуют дисплазии I и II степени, а ЦИН 3 включает дисплазию III степени и рак in situ (рис. 3).
В структуре патологии шейки матки у женщин репродуктивного возраста ЦИН составляют от 17 до 29%. По данным ВОЗ (2008) распространенность в мире ЦИН 1 степени составляет 30 млн случаев в год, а ЦИН 2–3-й степени – 10 млн. Средний возраст пациенток с диагностированным ЦИН всех степеней составляет 34,0±3,0 года. Анализ исходов ЦИН показал, что при ЦИН 1 регрессия наблюдается в 57% случаев, персистенция – в 32%, прогрессия – в 11%, а развитие РШМ – только в 1% случаев. В то время как при выявлении ЦИН 3 малигнизация происходит более чем в 12% случаев, а регрессия – лишь в 32% (таблица) [5].
Известно, что для снижения показателя заболеваемости РШМ необходимы профилактические мероприятия. При этом первичная профилактика РШМ предполагает проведение мероприятий в отношении лиц, в том числе не имеющих признаков заболевания на момент обследования, с целью предотвращения его развития в дальнейшем. В последнее время наиболее перспективной считается вакцинация против вируса папилломы человека (ВПЧ). Вторичная профилактика РШМ предполагает раннее выявление и последующее лечение предраковых изменений шейки матки.
В настоящее время в нашей стране функционирует в основном система оппортунистического скрининга, при которой обследуются женщины, обратившиеся за помощью в лечебно-профилактические учреждения. В то время как в некоторых странах за рубежом широко внедрена система организованного скрининга. При таком подходе определяется популяция женщин, подлежащих скринингу, и его периодичность. Кроме того, женщины активно приглашаются на обследование. Данные современной литературы свидительствуют о более высокой эффективности организованного скрининга по сравнению с оппортунистическим [5].
Внедрение скрининга в современную медицинскую практику в России могло бы открыть широкие возможности для профилактики и диагностики предрака и РШМ и явиться основой для снижения заболеваемости в целом сохранения репродуктивного здоровья женщины.
В связи с этим актуальной проблемой является поиск маркеров для раннего выявления и прогнозирования прогрессии ЦИН и РШМ, которые позволили бы усовершенствовать подходы к прогнозу течения заболевания и эффективности терапии. Кроме того, необходимо выявить пациенток высокого риска и на основе найденных критериев определить группу женщин, требующих более тщательного наблюдения.
В последнее время, при поиске молекулярных и функциональных биомаркеров РШМ научными коллективами разных стран уделяется пристальное внимание состоянию митохондриального аппарата клеток эпителия шейки матки для установления причинно-следственных связей между метаболическими нарушениями и их дальнейшим развитием [6, 7]. Изучение особенностей метаболических реакций в клетках имеет значение для понимания механизмов возникновения и развития ЦИН и РШМ. Отличительным свойством раковых клеток является состояние их метаболического репрограммирования, на начальной стадии возникновения злокачественного новообразования играющего роль патогенетического триггера. При этом сформировавшийся тип метаболизма, так называемый аэробный гликолиз, характерен для всех стадий канцерогенеза, определяя выраженность всех признаков, присущих опухолевым клеткам [8].

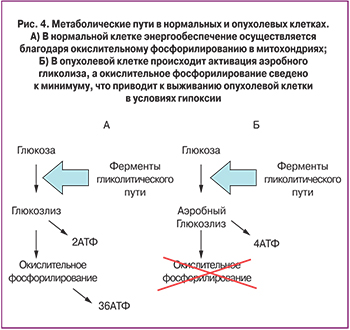
Известно, что глюкоза является одним из основных источников энергии для большинства клеток. В норме глюкоза поступает в цитоплазму клетки с помощью интегральных белков – переносчиков семейства GLUT 1-4, локализованных в плазмалемме [9]. В цитоплазме глюкоза претерпевает ряд последовательных преобразований структуры в гликолитическом пути, превращаясь в пировиноградную кислоту. В отсутствие кислорода при гипоксии, характерной для растущего новообразования [10], пируват превращается в лактат (молочную кислоту) с одновременной регенерацией НАДН (никотинамидадениндинуклеотид восстановленный). При этом в клетках опухоли экспрессируется переносчик лактата, обеспечивающий выброс этого соединения из клетки в межклеточное пространство с последующим поглощением его соседними клетками с аэробным метаболизмом. В присутствии кислорода в нормальной ткани пируват транспортируется в митохондрии, где после декарбоксилирования превращается в ацетил-КоА, окисляющийся в цикл трикарбоновых кислот до углекислого газа и воды. Образующиеся при этом НАДН и ФАДН2 (флавинадениндинуклеотид восстановленный) используются как источники электронов в дыхательной цепи, обеспечивая формирование трансмембранного протонного градиента, конвертируемого АТФ-синтетазой в энергию связи при образовании молекулы АТФ (аденозинтрифосфорной кислоты). При окислении 1 молекулы глюкозы до углекислого газа и воды об разуется 38 молекул АТФ, при этом нормальные клетки используют процессы тканевого дыхания и окислительного фосфорилирования для получения 90% общей продукции АТФ, и только 10% образуется в ходе анаэробного гликолиза [11].
Метаболическое репрограммирование раковой клетки характеризуется сдвигом в энергообеспечении клетки от митохондриального окислительного фосфорилирования к аэробному гликолизу и является важнейшей отличительной характеристикой, которую приобретают клетки еще в процессе опухолевой трансформации [8]. В двадцатых годах прошлого столения Отто Варбург показал, что в опухолевых клетках повышен уровень гликолиза с образованием лактата даже в условиях достаточного количества кислорода – так называемый эффект Варбурга [12]. Варбург считал, что аэробный гликолиз является результатом нарушения окислительного метаболизма в митохондриях [13]. На сегодняшний день считают, что аэробный гликолиз присущ раковым клеткам уже на самых ранних стадиях канцерогенеза, еще до момента, когда клетки начинают испытывать недостаток кислорода в отсутствие васкуляризации быстро растущей опухоли. Готовность к быстрому захвату глюкозы, выбросу лактата и высокая скорость аэробного гликолиза, опосредованные повышенной экспрессией соответствующих переносчиков и ферментов, дают опухолевым клеткам преимущества перед окружающими опухоль нормальными клетками, обеспечивая их выживание в условиях рано или поздно развивающейся гипоксии и нарастания концентрации проапоптотических цитокинов при росте опухоли [14].
На сегодняшний день показано, что помимо переносчиков и ферментов, принимающих участие в метаболических процессах, в клетках опухоли нарастает концентрация полипептидов регуляторного типа, индуцируемых гипоксией, в том числе белков семейства HIF (hypoxia inducible factor – гипоксия-индуцируемый фактор). α и β-субъединицы HIF формируют гетеродимер, проникающий в ядро и выполняющий роль фактора транскрипции при связывании с соответствующим участком хромосомальной ДНК. При этом происходит активация ряда генов, в том числе контролирующих биогенез митохондрий, приводящая к изменению уровня экспресии соответствующих белков. По данным исследований последних лет выявлен высокий уровень содержания белка HIF-1α в биопсиях опухолей молочной железы, эндометрия, пищевода, желудка, легких, полости рта, гортани, яичника, поджелудочной железы, кишечника, а так же шейки матки, что расценивается как плохой прогностический фактор в отношении общей выживаемости [15]. В ряде работ на животных моделях было экспериментально показано, что снижение уровня экспрессии HIF-1α в злокачественных опухолях достоверно замедляет прогрессию и метастазирование опухоли [16, 17]. Получены данные, свидительствующие о том, что увеличение уровня экспрессии HIF-1α считается предиктором плохого ответа на химиотерапию у больных раком молочной железы [18, 19]. По другим данным увеличение уровня экспрессии HIF-1α также является предиктором рецидива рака предстательной железы и кишечника [20].
X. Tang и соавт. в своих работах показали, что экспрессия HIF-1α при ЦИН коррелирует со стадией заболевания. Так при ЦИН 1-2 наблюдалась незначительная экспресссия изучаемого маркера, а при ЦИН 3 отмечалась гиперэкспрессия, в то время как в нормальных клетках эпителия шейки матки увеличение экспрессии HIF-1α не наблюдалось [21].
При этом высокая экспрессия HIF-1α является плохим прогностическим фактором у больных раком молочной железы, независимо от степени поражения лимфатических узлов, хотя считается, что пациентки с непораженными лимфатическими узлами имеют лучший прогноз [20]. Однако не все авторы обнаруживают связь между экспрессией HIF-1α и прогнозом. Так, A. Mayer и соавт., изучая экспрессию HIF-1α при РШМ, не выявили четкой корреляции между содержанием этого белка и стадией заболевания, частотой метастазирования опухоли, возникновением рецидивов заболевания и выживаемостью больных и сделали вывод, что HIF-1α нельзя рассматривать в качестве прогностически значимого маркера РШМ [22]. Наконец, в рамках экспериментальной работы о динамике белков в клетках растущей опухоли были получены данные о повышении экспрессии HIF-lα только в опухолевых клетках, тогда как в предопухолевых клетках подобного явления не наблюдалось [23].
Таким образом, можно предположить, что хотя гипоксия является важным фактором индукции и прогрессии РШМ, значение этого явления в канцерогенезе и его вклад в метаболическое репрограммирование клеток до конца не ясны. Ранее считалось, что гипоксия – это патологическое состояние, характеризующееся недостатком энергообеспечения клетки, опосредованное менее эффективным метаболизмом. В настоящее время исследователи склоняются к мнению, что гипоксия – это иное физиологическое состояние клетки, поддерживающее низкий уровень клеточной дифференцировки – с одной стороны благоприятствующее пролиферации и регенерации тканей в онтогенезе, с другой стороны, при определенных условиях способствующее неопластической трансформации клеток и росту опухоли.

In vitro клетки, подвергнутые экспериментальной гипоксии, демонстрируют способность к увеличению скорости пролиферации, остановке дифференцировки, увеличению способности к миграции и инвазии, а так же устойчивости к генотоксическому стрессу и проапоптогенам. При этом происходит резкий подъем внутриклеточного содержания продуктов гликолиза, в генезе которого выделяют ряд стадий – изменение скорости транспорта глюкозы, увеличение скорости вовлечения глюкозы в гликолиз на фоне ингибирования ферментов глюконеогенеза и более эффективный экспорт лактата за пределы клетки (рис. 4) [24, 25].
Глюкоза поступает в клетку с помощью глюкозных переносчиков GLUT 1-12. По данным литературы в опухолевых клетках повышено содержание GLUT 1, 3 и 12, причем в тех клетках, которые испытывают гипоксию, существенно возрастает роль GLUT-1 [9, 26]. По данным литературы белок GLUT-1 обнаруживается на сходном уровне в нормальных клетках и в клетках доброкачественных опухолей, в то время как в злокачественных опухолях, в том числе при РШМ, наблюдается значительное повышение его экспрессии. Существуют данные, что увеличение экспрессии GLUT1 коррелирует с пролиферативной активностью клеток опухоли и с ее более агрессивным поведением [27]. Группа ученых под руководством R. Airley в своем исследование выявила статистически значимую корреляцию между уровнем экспрессии GLUT-1 и стадией цервикального канцерогенеза [28]. Под руководством Markowska была оценена роль и прогностическая значимость маркера GLUT1 в канцерогенезе шейки матки, а так же проанализирован уровень экспрессии данного маркера и выявлена корреляция между уровнем его экспрессии и стадией заболевания. В исследование вошло 106 женщин с различными стадиями РШМ, которым проводилась биопсия шейки матки. Уровень экспрессии изучаемого белка определялся иммуногистохимическим методом с применением антител к GLUT-1 и наибольшие величины были зарегистрированы для низкодифференцированных клеток РШМ. Кроме того, было показано что показатель пятилетней выживаемости у женщин с высоким уровнем экспрессии GLUT-1 значимо ниже, чем в группе женщин с такой же стадией заболевания, но низким уровнем экспрессии изучаемого маркера. Исследователи пришли к выводу что в диагностике РШМ, в том числе при его прогрессии, GLUT-1 можно рассматривать в качестве прогностически значимого маркера [29]. Хотя эти результаты были впоследствии всопроизведены и подтверждены рядом работ [30, 31], в литературе существуют и противоречащие им данные о низкой прогностической ценности GLUT-1, но проведенные исследования немногочисленны и отличаются небольшой выборкой, что не позволяет использовать их в качестве источников для анализа о применимости оценки уровня GLUT-1 в диагностике РШМ [32, 33]. В связи с этим мнение большинства исследователей склоняется к признанию данного белка в качестве прогностически значимого маркера для раннего выявления ЦИН и РШМ.
Метаболическое превращение поступившей в клетки глюкозы начинаются с ее фосфорилирования гексокиназой (ГК) (АТФ: D-гексозо-6-фосфотрансферазой, ЕС 2.7.1.1.), имеющей несколько изозимов, кодируемых различными генами и имеющими различную тканеспецифичность и внутриклеточную локализацию. В настоящее время вопрос о том, по какому механизму у высших эукариот активация гликолиза приводит к подавлению окислительного фосфорилирования до конца неясен. Существует теория, что метаболическое репрограммирование возникает как адаптивная реакция в ответ на гипоксию еще на ранних этапах канцерогенеза при активации гликолиза в клетке. Имеются работы, которые показывают, что активация гликолиза в опухолевых клетках ассоциирована со злокачественностью опухоли, устойчивостью клеток опухоли к химио- и лучевой терапии, и как следствие, плохим прогнозом [34, 35].
Кроме того известно, что изозим ГК-2 редко встречается в нормальных клетках различных тканей (за исключением скелетной и сердечной мышцы, почечной мезангиальной и жировой ткани), однако его экспрессия резко повышается при различных онкологических заболеваниях, включая рак желудка, глиобластому, гепатоцеллюляоную карциному, рак молочной железы и рак гортани [36–42]. Недавно были получены данные об увеличении экспрессии данного белка при РШМ. Исследователи показали, что уровень экспрессии ГK-2 в группе с диагнозом РШМ был гораздо выше, чем в контрольной группе [43].
 Нами было показано, что подобное увеличение уровня экспрессии ГК-2 происходит и на ранних стадиях образования, и на стадии прогрессии неоплазий, в частности на стадии ЦИН1-3. Кроме того, обнаружена тенденция к увеличению уровня экспрессии ГK-2 в соответствии со степенью поражения шейки матки [44]. На основании полученных данных, можно предположить существование тесной связи между уровнем экспрессии ГK-2 и степенью поражения шейки матки, в связи с чем данный белок можно рассматривать в качестве биологического маркера прогрессии диспластических процессов и развития РШМ. Недавно было показано, что выживаемость пациентов отрицательно коррелирует с уровнем экспрессии ГK-2 при гепатоцеллюлярной карциноме и раке молочной железы [39, 41]. К сожаления, число подобных исследований в отношении РШМ пока невелико.
Нами было показано, что подобное увеличение уровня экспрессии ГК-2 происходит и на ранних стадиях образования, и на стадии прогрессии неоплазий, в частности на стадии ЦИН1-3. Кроме того, обнаружена тенденция к увеличению уровня экспрессии ГK-2 в соответствии со степенью поражения шейки матки [44]. На основании полученных данных, можно предположить существование тесной связи между уровнем экспрессии ГK-2 и степенью поражения шейки матки, в связи с чем данный белок можно рассматривать в качестве биологического маркера прогрессии диспластических процессов и развития РШМ. Недавно было показано, что выживаемость пациентов отрицательно коррелирует с уровнем экспрессии ГK-2 при гепатоцеллюлярной карциноме и раке молочной железы [39, 41]. К сожаления, число подобных исследований в отношении РШМ пока невелико.
В настоящее время появляются данные, говорящие в пользу того, что в опухолевых клетках ГК-2 участвует не только в обеспечении выживания их во время метаболического репрограммирования, но и влияет на чувствительность клеток к проапптогенным факторам [45, 46]. Белок р53 является продуктом гена-супрессора опухоли р53 и экспрессируется во всех клетках организма, запуская транскрипцию группы генов. Известно, что данный фактор транскрипции активируется при накоплении повреждений в ядерной ДНК. При этом, в отсутствие повреждений генетического аппарата белок р53 находится в неактивном состоянии, а при появлении повреждений ДНК активируется. Результатом активации р53 является остановка клеточного цикла и репликации ДНК, а так же запуск апоптоза. Важным этапом каскада апоптотических реакций в клетке является именение проницаемости митохондриальной мембраны для проапоптогенов межмебранного пространства, которое осуществляется за счет сдвига баланса взаимодействующих белков семейства Bcl-2. Интересно, что среди представителей этого семейства антиапоптозные белки (Bcl-2, Bcl-Xl, Bcl-Wl) действуют в сторону понижения проницаемости митохондриальной мембраны для цитохрома с, в то время как проапоптозные белки (Bax, Bid, Bim, BNIP и др.), наоборот, стимулируют выброс из межмебранного пространства митохондрий цитохрома с, что и является ключевым актом активации апоптического каскада. При этом белок р53 влияет как на внешний (опосредованный лигандами Apo/Trail), так и на митохондриальный пути индукции апоптоза [47]. Действуя на митохондриальный путь апоптоза, р53 подавляет транскрипцию анти-апоптозных белков Bcl-2 и активирует транскрипцию про-апоптозных белков. Механизм контроля проницаемости мембран для цитохрома с и других апоптогенов состоит в том, что проапоптозные белки взаимодействуют с внешней мембраной митохондрии, изменяя состояние белка VDAC1 (потенциал-зависимый анионный канал 1) в сторону повышения вероятности формирования проапоптотических протеолипидных структур, в то время как антиапоптозные белки препятствуют таким изменениям. Образующийся мультибелковый комплекс и обеспечивает выброс из межмебранного пространства митохондрий в цитозоль цитохрома с, активирующего апоптоз [48]. При этом происходит открытие гигантской митохондриальной поры, которая вызывает деполяризацию митохондрий, потерю пиридиновых нуклеотидов, разобщение окислительного фосфорилирования, набухание митохондрий и разрыв внешней мембраны [49].
Известно что ГК-2 способна к адсорбции на внешней мембране митохондрий и взаимодействию с белком VDAC1, ответственным за обмен метаболитами между цитозолем и митохондриями. В то же время, VDAC1, как уже упоминалось выше, непосредственно вовлечен в связывание на поверхности митохондрий проапоптотических белков Bax/Bid и антиапоптотического Bcl-2. Связывание проапоптогенов и ГK-2 носит конкурентный характер, при этом, фермент связывается с VDAC1 более прочно, поэтому повышение экспрессии ГК-2 в опухолевых клетках приводит к блокированию индукции апоптоза на самом первом этапе сборки проапоптогенного комплекса [50, 51]. Увеличение числа сайтов посадки ГK2, количество которой резко растет в опухолевой клетке, по-видимому, также приведет к сдвигу равновесия от возможности индукции апоптоза к его полному запрету.
Опубликованные результаты свидетельствуют об увеличении уровня экспрессии VDAC1 во многих видах рака и падении уровня экспрессии проапотогенов Bax, Bid. Методами масс-спектрометрии, иммуногистохимии и вестерн-блотт анализа было показано увеличение уровня экспрессии белков, участвующих в контроле апоптоза, в том числе и VDAC1, в клетках рака желчевыводящих путей и желудка [52, 53]. Изучены уровни экспрессии VDAC1 при раке щитовидной железы, легких, яичников, поджелудочной железы, при меланоме, глиобластом, а так же РШМ [43]. В другом экспериментальном исследовании показано увеличение уровня экспрессии VDAC1 в образцах ткани легкого, взятых из разных областей у одного и того же пациента с диагностированной опухолью и нормальной тканью легкого. Кроме того, наблюдалось не только увеличение уровня экспрессии VDAC1, но и корреляция уровня экспресии с опухолевой прогрессией и чувствительностью к химиотерапии [54].
Получены данные, которые говорят об увеличении экспрессии белка VDAC1 в периферической крови у больных с хроническим лимфолейкозом по сравнением с уровнем его экспрессии в крови здорового человека. Так при проведении Вестерн-блот-анализа клеток периферической крови пациентов с хроническим лимфолейкозом наблюдалось кратное увеличение уровня экспрессии VDAC1, по сравнению с клетками крови здоровых доноров [55]. Таким образом, VDAC1 может быть использован в качестве молекулярного маркера ранней диагностики, прогнозирования развития и течения рака, а так же для определения эффективности леченияя. Однако, хотя роль VDAC1 в канцерогенезе изучена достаточно для понимания значимости этого белка в прогрессии опухолевых неоплазий, роль этого белка в развитии ЦИН и РШМ начинает только изучаться. В частности нами недавно было впервые показано увеличение уровня экспрессии VDAC1 как для HSIL, предшествующих развитию РШМ, так и на ранних стадиях, в том числе и для ASCUS и LSIL [44].
В настоящее время активные формы кислорода (АФК), производимые митохондриями, рассматриваются в качестве одного из факторов, усиливающих внутриклеточный окислительный стресс. Повышенная генерация митохондриальных (мт)АФК при окислительном стрессе приводит к повреждениям белков, липидов, нуклеиновых кислот, а так же других биологических молекул и как в следствие к метаболическому репрограммированию клетки и развитию рака. Считается, что большинство повреждений ДНК удаляется белками эксцизионной репарации оснований (base excision repair, BER). BER устраняет небольшие повреждения, такие как окисленные или восстановленные азотистые основания, небольшие аддукты и повреждения, производимые метилирующими агентами. Ферменты, вовлеченные в ВЕR, кодируются полиморфными генами, аллельные варианты которых ассоциированы с разной функциональной активностью, что отражается на эффективности репарации. Доказано, что гуанин является одной из наиболее чувствительных и биологически важных мишеней при повреждении ДНК активными формами кислорода, а продуктом повреждения – 8-оксогуанин (8-OG). В настоящее время считается, что 8-оксогуанин является значимым биомаркером окислительного повреждения ДНК. В литературе имеются данные, показывающие что существует связь между образованием 8-OG и такими процессами как мутагенез, канцерогенез, старение и другими [56].
Удаление остатков 8-оксогуанина из ДНК человека осуществляет фермент 8-оксогуанин-ДНК-гликозилаза (hOGG1). Ген hOGG1 кодирует ключевой фермент эксцизионной репарации оснований, удаляющий из ДНК остатки 8-оксогуанина, образующегося под действием АФК. Ген hOGG1 картирован на хромосоме 3 [57]. Наиболее детально изучен полиморфизм экзона 3, который приводит к замене серина 326 на цистеин. По мнению авторов, именно эта замена играет важную роль в запуске патологического процесса [58, 59]. При изучении влияния этой замены на активность 8-оксогуанин-ДНК-гликозилазы при канцерогенезе получены неоднозначные результаты. Обнаружено, что понижение активности фермента у носителей варианта hOGG1-Cys326 приводит к снижению эффективности репарации окислительных повреждений ДНК и как следствие, к высокому риску возникновения рака легкого, желудка, карциномы яичников, пищевода, гортани и простаты у носителей Cys-аллели [60].
Показано, что риск развития рака зависит как от этнической принадлежности, так и от типа и локализации опухоли. Недавно выполненный мета-анализ продемонстрировал рисковую значимость полиморфизма hOGG1 Ser326Cys для различных типов рака в странах Азии. Однако, наиболее устойчивая ассоциация замены Ser → Cys с опухолями легкого наблюдалась не только у азиатов, но и у европейцев. Этот же вариант гена hOGG1 оказался нейтральным по отношению к раку мочевого пузыря независимо от этнической принадлежности обследуемых, но, согласно другим авторам, является фактором риска развития рака мочевого пузыря в японской популяции и у населения Северной Индии [61].
Получены предварительные данные исследования о роли полиморфизмов гена hOGG1 при РШМ. Было показано, что нет значимой разницы распространенности полиморфизма гена hOGG1 Ser326Cys между нормальными клетками и клетками РШМ. Однако, имеющиеся публикации малочисленны и описанные исследования выполнены на небольшой выборке, что не позволяют сделать окончательный вывод о роли полиморфизмов гена hOGG1 в развитии РШМ.
На данный момент общепризнано, что укорочение теломер ассоциированно с развитием рака и, возможно, является предрасполагающим фактором для развития ряда онкологических заболеваний, в том числе и для РШМ [62]. Теломеры – это структурные комплексные компоненты хромосом, состоящие из терминальных повторяющихся последовательностей ДНК, связанных с белками теломерного комплекса. Теломеры расположены на концах каждой хромосомы и выполняют стабилизирующую и защитную функции. Теломеразы состоят из двух основных компонентов: теломеразная обратная транскриптаза – TERT (наиболее важный домен-hTERT каталитическая субъединица) и TER-специальная теломеразная РНК. Одна из основных функций теломер является защита генетической информации хромосом при делении клеток. Критически короткие теломеры неспособны защищать хромосомы от повреждения при митозе (деление клетки). Их появление является сигналом для выхода клеток из митотического цикла. При укорочении теломеры в клетке происходит резкое изменение матаболизма, и в первую очередь нарушение репликации ДНК, которое запускает механизмы клеточного старения и гибели. Теломеразная активность в нормальных соматических клетках подавлена. Однако, исключением являются лимфоциты, клетки высокопролиферативных тканей (таких как костный мозг и др.) и пролиферативные клетки возобновляемых тканей (такие как клетки крипты кишки, базального слоя кожи и эпителиальные клетки шейки матки), а также гормонально зависимые клетки молочной железы и эндометрия [63].
Любопытно, что фермент ТЕRТ обнаруживается в митохондриях, где для данного фермента отсутствует субстрат, поскольку для мтДНК на данный момент наличие теломер не показано. Оказалось, что основная функции ТЕRТ в митохондриях – это стабилизация мтДНК и ее защита от повреждений. Более того, ТЕRТ, взаимодействуя с кодирующими участками мтДНК регулирует активность генов и принимает участие в репликации и репарации мтДНК, а также по неизвестному механизму подавляет продукцию в митохондриях АФК, что уменьшает вероятность апоптоза и некроза при окислительном стрессе [64].
Известно, что клетки большинства злокачественных опухолей характеризуются высокой активностью теломеразы, которая поддерживает длину теломер на постоянном уровне. Это дает им способность делиться неограниченное число раз, что вызывает нестабильность генома. При нестабильности генома высока вероятность спонтанных хромосомальных аббераций, начиная от количественных изменений и заканчивая структурными аномалиями. В большенстве злокачественных клеток наблюдается постоянная высокая экспрессия генов теломеразы, что поддерживает длину теломер на уровне, необходимом для их функционирования и быстрой пролиферации клеток опухоли [65].
В заключение важно отметить, что недостаток кислорода является общей чертой многих солидных опухолей, что позволяет им выживать в неблагоприятных условиях. При недостатке кислорода изменяются многие аспекты клеточного метаболизма. Гипоксия инициирует адаптационные процессы, которые могут быть выгодны для опухолевой прогрессии, происходит переключение клеточного дыхания с митохондриального окислительного фосфорилирования на анаэробный гликолиз. В ответ на гипоксию и подъем уровня HIF-1α повышаетсяе уровень экспрессии GLUT-1 и синтеза продуктов гликолиза, а также фермента ГK-2, который связываясь с VDAC1 на митохондриальной мембране, блокирует индукцию апоптоза в опухолевых клетках. В ответ на резкое изменение метаболизма в клетке нарастает нестабильность генома, наблюдается высокая экспрессия генов ТЕRТ для поддержания длины теломер на уровне, необходимом для их функционирования и быстрой пролиферации опухолевой клетки. Вследствие данных изменений клетка переходит в низкодифференцированное состояние, индуцируется рост и прогрессия опухоли, повышается ее способность к инвазии и метастазированию.
Заключение
Таким образом, метаболическое репрограммирование, вызываемое падением репаративных возможностей клетки в отношении поврежденной ДНК, является отличительной чертой раковых клеток и играет важную роль в адаптации и выживанию в новых условиях. При этом, репрограммирование играет роль триггера индукции, развития и прогрессии злокачественных новообразований, в том числе при ЦИН и РШМ. Несмотря на то, что метаболическая перестройка в раковых клетках довольно сложный процесс, и детальные механизмы, лежащие в его основе, остается еще уточнить, результаты исследований особенностей метаболизма предраковых и раковых клеток будут важным не только для более ранней и совершенной диагностики ЦИН и РШМ, но и для формирования тактики ведения таких больных, контроля эффективности за проводимым лечением и определения риска вторичной неопластической трансформации.


